La recherche clinique concerne l’être humain. Elle a pour objectifs d’améliorer la connaissance de la maladie, améliorer la qualité de vie, trouver de nouvelles méthodes d’exploration et quantification de la maladie ou de traitements, et développer des thérapies.
Il existe des études et des essais cliniques comme présenté dans le tableau ci-dessous (traduit de FARA : Clinical Trials 101 : A Guide For Participants)
ETUDES CLINIQUES
C’est une étude d’observation.
- Objectifs : décrire/mesurer/observer la maladie.
- Aucune intervention n’est testée (aucun médicament, aucun traitement, aucun dispositif).
- Exemples : études de biomarqueurs, études d’histoire naturelle, d’imagerie, etc.
- Le consentement éclairé s’applique dans certains cas.
ESSAIS CLINIQUES
C’est une intervention.
- Objectifs : tester un médicament, un traitement, un dispositif.
- En général, un patient ne peut pas participer à plus d’un essai clinique à la fois.
- Le consentement libre, éclairé et écrit s’applique.
Les études cliniques
Le consortium européen Effacts
Regroupant chercheurs fondamentaux et cliniciens, spécialistes de l’ataxie de Friedreich, EFACTS (European Friedreich Ataxia Consortium for Translational Studies) a été créé en 2010. Le Consortium EFACTS est un réseau de structures hospitalières et de recherche (16 en octobre 2022) couvrant plusieurs pays européens (10 en octobre 2022) pour collecter des données longitudinales (= sur la durée) sur l’histoire naturelle d’une grande cohorte de patients génétiquement définis comme atteints d’ataxie de Friedreich. A la direction du consortium, l’hôpital universitaire d’Aix-la-Chapelle (Pr Jörg Schulz) qui a succédé au Prof. Massimo PANDOLFO.
Les objectifs sont les suivants :
- Mieux comprendre l’histoire naturelle de l’Ataxie de Friedreich, c’est-à-dire mieux comprendre tous les aspects de la maladie avec sa complexité
- Etudier la structure et la fonction de la frataxine
- Etudier la pathogénicité dans la maladie
- Etudier les mécanismes de répression du gène de la frataxine
- Créer de nouveaux modèles cellulaires et animaux pour l’étude de la maladie
- Découvrir des biomarqueurs (très importants pour les essais cliniques)
- Identifier les modificateurs génétiques de la maladie
- Développer de nouvelles approches thérapeutiques.
La banque de prélèvements biologiques (bio-repository) et le registre d’histoire naturelle (1.103 patients en octobre 2022 et 67 sujets contrôle sains), accessibles à la communauté scientifique, permettent les études sur l’histoire naturelle de la maladie (symptômes, développement, évolution…) ainsi que le recrutement de patients pour les essais cliniques. Les premiers patients enrôlés sont suivis depuis plus de 10 ans.Des liens de plus en plus forts ayant été établis avec FARA aux Etats-Unis, une puissante association US de soutien à la recherche pour l’ataxie de Friedreich, des négociations ont été menées depuis plusieurs années qui ont abouti en 2022 à la signature d’une convention pour organiser le partage des données européennes et américaines. Cette convention institutionnalise la constitution d’une plate-forme neutre et indépendante de collaboration et d’analyse de données pour promouvoir le partage de données essentielles sur l’ataxie de Friedreich, selon un référentiel de procédures commun à toutes les structures parties prenantes.
Documents associés
Fonctionnement des essais cliniques
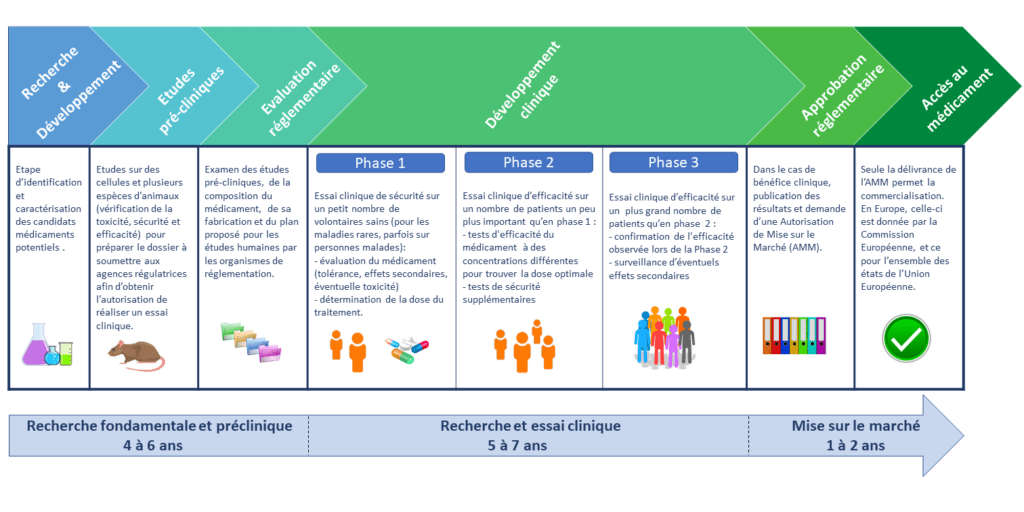
Les étapes d'un essai clinique
Etudes précliniques : le candidat médicament est testé sur des cellules et des animaux (non BPL). Le promoteur doit tester le nouveau candidat médicament sur des animaux pour vérifier la toxicité (BPL). Plusieurs espèces sont utilisées pour recueillir des informations sur la sécurité et l’efficacité du composé. Ces études servent à préparer le dossier à soumettre aux agences régulatrices afin d’obtenir l’autorisation pour réaliser un essai clinique.
Evaluation réglementaire : les organismes de réglementation examinent les études pré-cliniques, la composition du médicament, sa fabrication et le plan proposé pour les études humaines. Pour réaliser un essai clinique aux USA, le promoteur doit soumettre une “IND” (Investigational New Drug Application, équivalent à la demande d’autorisation d’essai clinique auprès de l’ANSM). La FDA (Food and Drug Administration) examine la demande pour s’assurer que les études proposées, n’exposent pas les sujets humains à un risque déraisonnable de préjudice. La FDA vérifie également le consentement éclairé et la protection des sujets humains. En France, c’est le rôle des CPP (Comités de Protection des Personnes) et de l’ANSM (Agence Nationale de Sécurité du Médicament et des produits de santé). Essai clinique de sécurité chez l’homme. Il sert à évaluer si le médicament est sûr et s’il y a des effets secondaires indésirables.
Développement clinique : il se déroule en plusieurs phases.
Approbation réglementaire : s’il y a un bénéfice clinique, les développeurs demandent une autorisation de mise sur le marché (AMM). Le dossier regroupe les données des essais précliniques, cliniques, sur la qualité chimique ou microbiologique du produit fini et les procédés de fabrication de la substance active et du produit fini. Le dossier d’AMM aura le même contenu quelle que soit la procédure engagée : pour l’EMA (Europe) ou pour la FDA (NDA: New Drug Application). Dans le cas des maladies neurodégénératives et maladies rares, une procédure au niveau européen “centralisée” est obligatoire.
Accès au médicament : pour l’Europe, l’EMA formule une recommandation à l’intention de la Commission Européenne, qui prend ensuite une décision finale sur l’AMM sur le marché du médicament dans l’UE. L’AMM est donc délivrée par la Commission Européenne et est valable pour l’ensemble des Etats membres. L’AMM constitue un préalable obligatoire à toute possibilité de commercialisation d’une spécialité pharmaceutique. En France, elle est également indispensable avant la demande d’inscription au remboursement par l’Assurance Maladie.
Essais cliniques de Phase 4 : après l’approbation d’un médicament par les autorités de réglementation, il peut rester des questions sans réponse sur l’efficacité ou le profil de sécurité du médicament. Par conséquent, des études de Ph4 peuvent être menées pour répondre à ces questions, même après qu’un médicament a été approuvé par un organisme de réglementation et est disponible sur ordonnance.
Participer à un essai ou une étude clinique
Participer à un essai clinique
Un essai clinique sur un médicament consiste à étudier, selon un protocole précis, les effets d’une molécule chez l’homme, qu’il s’agisse de volontaires malades ou sains. Le but des essais est d’établir la sécurité d’emploi et l’efficacité d’une substance.
Les essais cliniques sont encadrés par des lois et doivent obéir aux règles de bonnes pratiques cliniques.
La participation à un essai est une démarche individuelle, volontaire et qui demande un engagement éclairé. Un essai clinique se fait sous la conduite d’équipes de médecins (appelés investigateurs), compétentes et expérimentées, qui connaissent avec précision la maladie, les traitements existants et les conditions requises pour participer à l’essai.
Le médecin doit vous expliquer l’essai auquel il vous propose de participer. Seul l’investigateur de l’essai clinique décide ou non d’accepter une candidature.
Documents associés
Principaux essais et études cliniques en cours
Les essais en cours
Les essais en cours - recrutement terminé
- LX2006 Ph1/2 thérapie génique – Promoteur Lexeo Therapeutics
- Identifiant ClinicalTrials.gov NCT05445323
- Notre compréhension du principe : Thérapie génique par virus adéno-associé (AAV) pour la cardiomyopathie: conçue pour porter le gène de la frataxine humaine aux cellules cardiaques. Pourrait atteindre le cœur, le muscle squelettique et d’autres tissus périphériques.
- Objectif : Thérapie génique pour la cardiomyopathie associée à l’ataxie de Friedreich
- Titre : Étude de Phase 1/2 sur la sécurité et l’efficacité de la thérapie génique LX2006 chez des participants atteints de cardiomyopathie associée à l’ataxie de Friedreich.
- Intervention / traitement : LX2006 (Vecteur viral adéno-associé codant pour le gène FXN [AAVrh.10hFXN]) à dose faible, dose moyenne et dose élevée
- Phase : 1/2
- Age de participation à l’étude : 18 à 50 ans
- Nombre de participants : 9
- Début de l’étude : 24 /08 /2022 (dernière mise à jour : 08/02/2024)
- Site et contacts : United States, California : Aaron Fisher 310-206-8153 adfisher@mednet.ucla.edu / Florida : Lucretia Campbell 813-974-5633 lcampbel@usf.edu / Iowa: Ciara Gibbs 319-384-9618 ciara-gibbs@uiowa.edu / Minnesota: Michaela Kolarova 507-266-7969 kolarova.michaela@mayo.edu / Pennsylvania : pas encore de recrutement / Canada, Quebec, Montreal : Antoine Duquette 514-890-8000 ext 30737 uit.eligibilite.chum@ssss.gouv.qc.ca
Les essais en cours de recrutement
- BIIB141 (Omaveloxolone) chez les enfants atteints d’ataxie de Friedreich (BOLD) – Promoteur : Biogen
- Identifiant ClinicalTrials.gov : NCT06054893
- Notre compréhension du principe : L’omaveloxone active la voie Nrf2, connue pour son rôle dans la réponse au stress oxydatif, et qui est réduite chez les patients atteints de l’ataxie de Friedreich. En France, la HAS a octroyé une autorisation d’accès précoce, après avis de l’ANSM concernant le bénéfice/risque présumé, pour SKYCLARYS® (omaveloxolone 50 mg, gélules) dans le traitement de l’ataxie de Friedreich (AF) chez les adultes et les adolescents âgés de 16 ans et plus.
- Objectif :
- Evaluer la pharmacocinétique, la sécurité et la tolérabilité de l’omaveloxolone après administration d’une dose unique dans 3 cohortes d’âge (≥2 à <6 ans, ≥6 à <12 ans et ≥12 à <16 ans).
- Evaluer la sécurité et la tolérance à long terme de l’omaveloxolone.
- Titre : Étude ouverte de Phase 1 visant à évaluer la pharmacocinétique, la sécurité, la tolérabilité, la pharmacodynamie et l’efficacité de l’omaveloxolone chez des enfants âgés de ≥2 à <16 ans atteints d’ataxie de Friedreich.
- Intervention / traitement :
- Partie A – Cohorte 1: patients âgés de ≥12 à <16 ans. Les patients recevront une dose orale unique de 150 mg d’omaveloxolone.
- Partie A – Cohorte 2: patients âgés de ≥12 à <16 ans. Les patients recevront une dose orale unique d’omaveloxolone à un niveau de dosage déterminé par une analyse Bayésienne de pharmacocinétique de population (popPK) utilisant les données de la Partie A – Cohorte 1 pour sélectionner la dose.
- Partie B: les patients âgés de ≥6 à <12 ans commenceront l’étude parallèlement à la partie A – Cohorte 2. Les patients recevront une dose orale unique d’omaveloxolone à un niveau de dosage déterminé par une analyse Bayésienne de pharmacocinétique de population (popPK) utilisant les données de la Partie A – Cohorte 1 pour sélectionner la dose.
- Partie C: les patients âgés de ≥2 à <6 ans recevront une dose orale unique d’omaveloxolone à un niveau de dosage déterminé par une analyse Bayésienne de pharmacocinétique de population (popPK) utilisant les données de la partie A et de la partie B pour sélectionner la dose.
- Phase : 1 pédiatrique
- Age de participation à l’étude : ≥2 à <16 ans
- Nombre de participants : 20
- Date de début de l’étude : 01/07/2024 (dernière mise à jour : 24/12/2024)
- Site et contacts : US Biogen Clinical Trial Center or Global Biogen Clinical Trial Center
Phone Number: 866-633-4636
Email: clinicaltrials@biogen.com
David Lynch, MD, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, United States, 19104 / Recruiting coordinator: 215-590-2242
- Nomlabofusp chez les adolescents et les enfants AF – Promoteur : Larimar Therapeutics
- Identifiant ClinicalTrials.gov : NCT06681766
- Notre compréhension du principe : Thérapie de remplacement de la frataxine, en utilisant le couplage de la frataxine à une protéine transporteuse.
- Objectif : Évaluer la sécurité et la tolérabilité du nomlabofusp (CTI-1601) chez les adolescents et les enfants atteints d’AF
- Titre : Étude de Phase 1 visant à évaluer la sécurité, la tolérabilité, la pharmacocinétique et la pharmacodynamique du Nomlabofusp sous-cutané chez les adolescents et les enfants atteints d’ataxie de Friedreich.
- Intervention / traitement : Il s’agit d’une étude randomisée, en double aveugle, contrôlée par placebo, évaluant une dose de nomlabofusp basée sur le poids versus un placébo chez des adolescents et des enfants atteints d’AF. Les participants recevront une injection sous-cutanée de 0,8 mg/kg, avec une dose maximale de 50 mg, une fois par jour pendant 7 jours, ou placebo. L’étude comprendra au moins deux cohortes avec au moins 12 à 15 participants dans chaque cohorte.
- Phase : 1
- Age de participation à l’étude : 2 à 17 ans
- Nombre de participants : 30
- Date de début de l’étude : 06/12/2024 (dernière mise à jour : 27/12/2024)
- Site et contacts : United States, Chevy Chase, Maryland, 20815; Tamanna Roshan Lal, MB ChB, 541-232-7818, troshanlal@UncommonCures.com ou Kerri Gallagher, RN, B, 267-234-2167, kgallagher@UncommonCures.com
- NAD+ et Exercice – Promoteur : Children’s Hospital of Philadelphia
- Identifiant ClinicalTrials.gov : NCT04192136
- Notre compréhension du principe : NAD+ est converti en NADH dans la cellule. Très important au sein de la mitochondrie pour produire de l’énergie. L’exercice augmente la masse musculaire, la capacité pulmonaire et cardiaque et améliore la fonction mitochondriale.
- Objectif : NAD+ et Exercice dans l’AF
- Titre : Supplémentation avec un précurseur NAD+ et entraînement physique pour augmenter la capacité aérobique dans l’ataxie de Friedreich
- Intervention / traitement : Nicotinamide Riboside (NR), Placebo, Exercice + NR ou Exercice + Placebo
- Phase : na
- Age de participation à l’étude : 10 à 40 ans
- Nombre de participants : 80
- Début de l’étude : 03 /09 /2020 (dernière mise à jour : 27/03/2024)
- Site et contacts : United States, Pennsylvania : Krisin L Wade 267-426-8724 wadekl@chop.edu
NAD : nicotinamide adénine dinucléotide / NADH : nicotinamide adénine dinucléotide réduit (coenzyme Q1)
- AAVrh.10hFXN Ph 1 – thérapie génique & prednisone – Promoteur : Weill Medical College of Cornell University
- Identifiant ClinicalTrials.gov : NCT05302271
- Notre compréhension du principe : AAVrh.10hFXN est un virus adéno-associé conçu pour porter le gène de la frataxine humaine aux cellules cardiaques.
- Objectif : Evaluer l’innocuité et l’efficacité préliminaire de l’AAVrh.10hFXN pour traiter la cardiomyopathie associée à l’ataxie de Friedreich
- Titre : Étude de phase 1a de thérapie génique avec AAVrh.10hFXN pour la cardiomyopathie de l’ataxie de Friedreich
- Intervention / traitement : AAVrh.10hFXN administré par voie intraveineuse et Prednisone (tous les participants suivront un traitement immunosuppresseur à base de prednisone pendant un total de 14 semaines).
- Phase : 1, ouverte, d’escalade de dose
- Age de participation à l’étude : 18 à 50 ans
- Nombre de participants : 25
- Début de l’étude : 22/02/2022 (dernière mise à jour : 17/10/2024)
- Site et contacts : United States, New York: Haley Bowe 646-962-2672 hab4007@med.cornell.edu ou Noor Hasan: 646-962-5583 noh4004@med.cornell.edu
- Entraînement respiratoire dans l’ataxie de Friedreich – Promoteur : University of Florida
- Identifiant ClinicalTrials.gov : NCT06539598
- Notre compréhension du principe : Etude interventionnelle prospective de patients atteints de l’AF qui reçoivent un entraînement de la force respiratoire pendant 12 semaines avec 2 visites au début et à la fin de l’étude. Les visites comprennent une évaluation de la déglutition par endoscopie à fibre optique, des tests de la fonction pulmonaire, une électromyographie de surface et des enquêtes auprès des patients.
- Objectif : Evaluer l’impact de l’entraînement respiratoire sur la déglutition et la fonction respiratoire.
- Titre : Impact de l’entraînement respiratoire sur la déglutition et la fonction respiratoire chez les patients atteints de l’ataxie de Friedreich.
- Intervention / traitement : Pas de traitement. Entraînement de la force respiratoire: Les participants recevront un appareil d’entraînement de la force respiratoire à emporter chez eux et à utiliser jusqu’à 5 fois par semaine pendant 12 semaines.
- Phase : N/A
- Age de participation à l’étude : 18 à 65 ans
- Nombre de participants : 8
- Début de l’étude : 29/08/2024 (dernière mise à jour : 19/09/2024)
- Site et contacts : Gainesville, Florida, United States, 32610
Clinical Research Center
Julia Prascak, BS
352-273-6855 juliaprascak@ufl.edu
Les essais non encore ouverts au recrutement
- CPCAF – Promoteur : Institut National de la Santé et de la Recherche Médicale (INSERM), France
- Identifiant ClinicalTrials.gov : NCT05874388
- Notre compréhension du principe : Batterie de tests conçus pour répondre aux besoins spécifiques des patients atteints d’AF, qui quantifient les aspects les plus importants des fonctions motrices, cognitives et de la parole.
- Objectif : Caractérisation du profil cognitif des patients atteints d’ataxie de Friedreich
- Titre : Caractérisation du profil cognitif des patients atteints d’ataxie de Friedreich
- Intervention / traitement : Pas de traitement / Etude comparative ouverte. L’étude comportera deux groupes : un groupe d’adolescents et d’adultes symptomatiques de l’AF avec un diagnostic moléculaire confirmé, suivi dans le service de génétique de l’hôpital Necker, et un groupe témoin de sujets sans aucune déficience motrice ou cognitive, recrutés parmi les parents sains des patients (frères et sœurs, cousins, etc.).
- Phase : na
- Age de participation à l’étude : 13 ans et +
- Nombre de participants : 70
- Date estimée de début de l’étude : 01/09/2023
- Site et contacts : Benoit Funalot, MD 01.49.81.28.60 benoit.funalot@aphp.fr
Documents associés
Avancer ensemble,
C’est aussi pouvoir compter sur votre aide !